
|
October 30, 2006 |
Computing Catches Up With Theory
Modeling the formation of blood clots
New computing tools have allowed Peter Richardson, professor of engineering and physiology at Brown University, to test ideas about blood flow and clotting that he first proposed more than 30 years ago. His collaboration with mathematics colleagues Igor Pivkin and George Karniadakis resulted in a model that integrates fluid dynamics with platelet biochemistry and could provide new insights into the treatment and prevention of strokes and heart attacks. | |||
|
Brown University Home |
PROVIDENCE, R.I. — Good science requires great patience. In many fields, ideas and theories surge ahead while the tools to test them can take decades to catch up. When Peter Richardson began talking with colleagues who were modeling blood flow through the vessels on the heart’s surface, he hardly suspected that the collaboration would lead to a test of ideas he had proposed more than 30 years before. The resulting model, described in the online edition of Proceedings of the National Academy of Sciences appearing the week of Oct. 30 to Nov. 3, could help evaluate candidate drugs aimed at preventing blood clots – a major cause of strokes, heart attacks and organ transplant rejection. 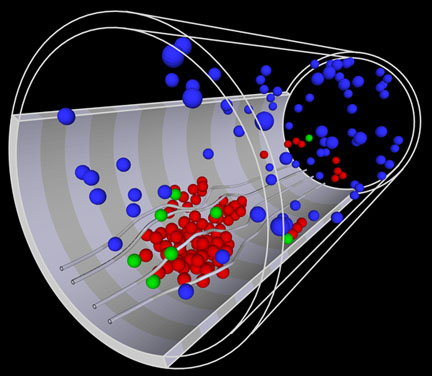 A model of platelets in action In 1970, Richardson, who had been working on the clotting problems associated with artificial organs, saw a paper describing the time course of clot formation in uninjured blood vessels. Gustav Born and his co-author, Nicola Begent, had found an odd relationship – shaped like a playground slide – between the rate of blood flow and the rate of blood clot, or thrombus, formation. As blood flow increased, the rate at which the clot grew increased rapidly, up to a point. After that point, the rate of growth declined suddenly and then gradually flattened out. Richardson recognized that there must be two groups of processes at work – probably one chemical and one physical. The increase in clotting with flow made sense. Faster blood flow meant more platelets encountered the clot each second, so more had a chance to be captured by it. But the decrease was puzzling. Researchers knew that ADP (adenosine diphosphate) released from injured tissue or existing thrombi could signal platelets to begin sticking together at the site of the injury. Richardson proposed that a short delay between triggering and activation could explain the decrease in aggregation with increased blood flow. “If triggering is the time when the alarm goes off,” said Richardson, a professor of engineering and physiology at Brown University, “then activation is when your feet hit the floor.” As blood flows faster, more platelets have already passed beyond the clot by the time they are ready for action. Richardson published his ideas at the time, but the computing power needed to simulate more than 50,000 platelets and their individual responses to a growing clot wasn’t yet available. Recently, when co-authors Igor Pivkin and George Karniadakis sought Richardson’s input on a similarly detailed simulation of blood flow in arteries on the heart’s surface, the team realized that the time had come to test Richardson’s hypothesis. Pivkin and Karniadakis modified their model to fit Richardson’s proposal and made hundreds of runs, changing flow rate, activation time, and the strength of attraction between activated platelets. Each set of runs produced a relationship between flow and aggregation remarkably similar to what Begent and Born had found in hamsters. While no recent experimental data set has the same detailed time course as Begent and Born’s, Richardson and colleagues have found that their model is able to reproduce several odd features of thrombus growth that have been observed both in the laboratory and in tissue pathologies from operating rooms and morgues. At high flow rates, they find the shape of the thrombus goes from “hill-like” to “carpet-like.” At very high flow rates and short activation times, they see secondary clots forming downstream from the primary site. The model is generalized in the sense that it does not require many specific details, such as the concentration of fibrinogen (which binds clots together) or the length of fibrinogen strands. It doesn’t need to specifically calculate concentration or diffusion of the activation factor. Yet it produces remarkably true-to-life results. In the next few years, the team will refine the model by incorporating molecular and sub-cellular details while enhancing the representation of fluid dynamics and interactions between platelets and other cells, working toward a truly multiscale model. Richardson also has physiological experiments planned that will allow more rigorous testing and refinement of the model. Revisiting this line of inquiry, says Richardson, may lead experimental researchers to explore drugs that could change the activation time of platelets, leading to better preventive strategies for clots, whether they result from blood vessel injury, atherosclerotic plaque or an artificial medical device. This work was supported by the National Science Foundation Interagency Modeling and Analysis Group, and computations were performed at the National Science Foundation supercomputing centers. ###### | |||