Next: Examples Up: Monte Carlo simulations Previous: Monte Carlo simulations
The Monte Carlo code needs the following files as an input
The parameters controlling the simulation are specified as command-line options. The first input parameter(s) needed by the code are the phase(s) whose thermodynamic properties are to be determined. There are two ways to invoke the Monte Carlo simulation code. When the command emc2 is used, a single Monte Carlo simulation is run to allow the calculation of thermodynamic properties of a single phase for the whole region of chemical potential and temperature where that phase is stable. The phase of interest is specified by a command-line option of the form
-gs=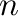 , , |
is a number between 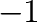 and
and 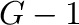 (inclusive), where
(inclusive), where 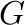 is the number of ground states.
The value indicates the disordered phase while values ranging from
is the number of ground states.
The value indicates the disordered phase while values ranging from 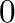 to indicate the phases associated
with each ground states ( denoting the ground state with the smallest composition).
When the command phb is used, two Monte Carlo simulations are run simultaneously
to enable the determination of the temperature-composition phase boundary associated with
a given two-phase equilibrium. The two phases are specified by
to indicate the phases associated
with each ground states ( denoting the ground state with the smallest composition).
When the command phb is used, two Monte Carlo simulations are run simultaneously
to enable the determination of the temperature-composition phase boundary associated with
a given two-phase equilibrium. The two phases are specified by
-gs1=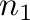 -gs2= -gs2=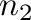 . . |
| -d1=directory 1 -d2=directory 2 |
The accuracy of the thermodynamic properties obtained from Monte-Carlo simulations is determined by two parameters: The size of the simulation cell and the duration of the simulation.
The size of the simulation cell is specified by providing the radius 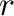 of a sphere
through the command-line option
of a sphere
through the command-line option
| -er=. |
![\includegraphics[width=1.0\textwidth]{mcparam}](img36.png) |
The duration of the simulations is automatically determined by the code from a user-specified target precision on the atomic composition of the phase, indicated by a command-line option of the form
-dx=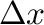 . . |
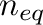 of
Monte Carlo steps the system is allowed to equilibrate before
thermodynamic averages are computed over a certain number
of
Monte Carlo steps the system is allowed to equilibrate before
thermodynamic averages are computed over a certain number 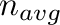 of Monte Carlo steps using the options
of Monte Carlo steps using the options
| -eq= -n=. |
The Monte Carlo code also needs additional parameters that specify which portion of a phase's free energy surface needs to be computed. With emc2, the range of temperatures to be scanned are specified in either one of the following two ways:
-T0=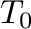 -T1= -T1=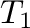 -dT= -dT=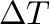 (for steps in direct temperature) (for steps in direct temperature) |
or -T0= -T1= -db=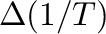 (for steps in reciprocal temperature). (for steps in reciprocal temperature).
|
) can be useful when
calculations are started from infinite temperatures down to a finite temperature.
The -T1 and -dT (or -db) options can be omitted if calculations at a single temperature
are desired. Since the program automatically stops when a phase transition is detected,
it is not necessary to know in advance the temperature range of stability of the phase. The
user only needs to ensure that the initial temperature lies within the region of stability
of the phase of interest. An obvious starting
point is 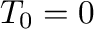 , since the ground state is then stable, by definition.
With the phb code, the syntax is
, since the ground state is then stable, by definition.
With the phb code, the syntax is
-T=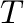 -dT= -dT= |
-k=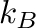 . . |
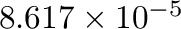 corresponds to energies
in eV and temperatures in Kelvin.
corresponds to energies
in eV and temperatures in Kelvin.
With emc2, the range of chemical potentials to be scanned needs to be specified.
Once again, only the starting point really matters, because the code will stop when a
phase transition is reached.
By default, chemical potentials are given in a dimensionless form, so as to
facilitate the link between the value of the chemical potential and the
phase that it stabilizes. For instance, a chemical potential equal to 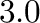 is
such that it would stabilize a two phase equilibrium between phase number
is
such that it would stabilize a two phase equilibrium between phase number
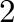 and phase number
and phase number 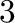 at absolute zero (see Figure 5.3b). A chemical potential between
and
at absolute zero (see Figure 5.3b). A chemical potential between
and 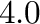 stabilizes phase number at absolute zero. While these ranges of
stability are no longer exact at finite temperature, this dimensionless chemical
potential still provides easy-to-interpret input parameters. The syntax is
stabilizes phase number at absolute zero. While these ranges of
stability are no longer exact at finite temperature, this dimensionless chemical
potential still provides easy-to-interpret input parameters. The syntax is
-mu0=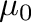 -mu1= -mu1=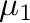 -dmu= -dmu=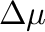 |
is the chemical potential step between each new simulation.
Chemical potentials
can also be entered in absolute value (say in eV, if the energies are in eV) by
specifying the -abs option.
Note that the output files always give the absolute chemical potentials, so that
thermodynamic quantities can be computed from them.
With phb, the initial chemical potential is optional when starting from absolute
zero because the code can determine the required value from the ground state energies.
It can nevertheless be specified (in absolute value) with the -mu=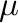 option, if a finite temperature starting point is desired.
option, if a finite temperature starting point is desired.
A list of the command line options of either the emc2 or phb codes can be displayed by simply typing either command by itself. More detailed help is displayed using the -h option.
[email protected] Wed, Dec 6, 2023 12:55:16 PM