- 1
-
A. van de Walle, S. Samanta, C. Nataraj, S. Zhu, H. Chen, H. Liu, and
R. Arroyave.
Revisiting the SGTE lattice stability of bcc aluminum.
Calphad Journal, 83:102628, 2023.
Download:
[SSRN]
[DOI]
[Cite]
.
- 2
-
H. Chen, S. Samanta, S. Zhu, H. Eckert, J. Schroers, S. Curtarolo, and
A. van de Walle.
Bayesian active machine learning for cluster expansion construction.
Computational Materials Science, 231:112571, 2024.
Download: [DOI]
[Cite]
.
- 3
-
S. Zhu, C. Nataraj, A. van de Walle, A. Perron, J. Shittu, J. Berry, J. T.
McKeown, and A. Samanta.
Probing phase stability in CrMoNbV using cluster expansion method,
CALPHAD calculations and experiments.
Acta Mater., 255:119062, 2023.
Download: [DOI]
[Cite]
.
- 4
-
S. Zhu, J. Schroers, S. Curtarolo, H. Eckert, and A. van de Walle.
Special glass structures for first principles studies of bulk
metallic glasses.
Acta Mater., accepted, 2023.
Download: [DOI]
[Cite]
[arXiv].
- 5
-
S. V. Ushakov, Q.-J. Hong, D. A. Gilbert, A. Navrotsky, and A. van de Walle.
Thorium and rare earth monoxides and related phases.
Materials, 16:1350, 2023.
Download: [DOI]
[Cite]
.
- 6
-
Q. Hong, A. van de Walle, S. V. Ushakov, and A. Navrotsky.
Integrating computational and experimental thermodynamics of
refractory materials at high temperature.
Calphad journal, 79:102500, 2022.
Download: [DOI]
[Cite]
.
- 7
-
H. Liu and A. van de Walle.
Rapid geometric screening of low-energy surfaces in crystals.
Symmetry, 14:2067, 2022.
Download: [DOI]
[Cite]
.
- 8
-
C. Oses, M. Esters, D. Hicks, S. Divilov, H. Eckert, R. Friedrich, M. J. Mehl,
A. Smolyanyuk, X. Campilongo, A. van de Walle, J. Schroers, A. G. Kusne,
I. Takeuchi, E. Zurek, M. Buongiorno Nardelli, M. Fornari, Y. Lederer,
O. Levy, C. Toher, and S. Curtarolo.
aflow++: a c++ framework for autonomous materials design.
Comput. Mat. Sci., 217:111889, 2022.
Download:
[arXiv]
[DOI]
[Cite]
Editor's choice.
- 9
-
Q.-J. Hong, S. V. Ushakov, A. van de Walle, and A. Navrotsky.
Melting temperature prediction using a graph neural network model:
from ancient minerals to new materials.
Proceedings of the National Academy of Science,
119:e2209630119, 2022.
Download: [DOI]
[Cite]
.
- 10
-
A. van de Walle, H. Chen, H. Liu, C. Nataraj, S. Samanta, S. Zhu, and
R. Arroyave.
Interactive exploration of high-dimensional phase diagrams.
JOM - J. Min. Met. Mat. S., 74:3478, 2022.
Download: [DOI]
[Cite]
Editor's choice.
- 11
-
E. Y. Cramer et al.
The united states covid-19 forecast hub dataset.
Scientific Data, 9:462, 2022.
Download: [DOI]
[Cite]
.
- 12
-
E. Y. Cramer et al.
Evaluation of individual and ensemble probabilistic forecasts of
covid-19 mortality in the US.
Proceedings of the National Academy of Science, 119:e211356111,
2022.
Download: [DOI]
[Cite]
[medRxiv].
- 13
-
C. Nataraj, E. J. L. Borda, A. van de Walle, and A. Samanta.
A systematic analysis of phase stability in refractory high entropy
alloys utilizing linear and non-linear cluster expansion models.
Acta Mater., 220:117269, 2021.
Download: [DOI]
[Cite]
[SSRN].
- 14
-
C. Nataraj, R. Sun, C. Woodward, and A. van de Walle.
First-principles study of the effect of Al and Hf impurities on
Co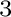 W antiphase boundary energies.
W antiphase boundary energies.
Acta Mater., 215:117075, 2021.
Download: [DOI]
[Cite]
.
- 15
-
R. Ojeda Mota, E. Lund, S. Sohn, D. Browne, D. Hofmann, S. Curtarolo, A. Van
de Walle, and J. Schroers.
Enhancing ductility in bulk metallic glasses by straining during
cooling.
Communications Materials, 2:23, 2021.
Download: [DOI]
[Cite]
.
- 16
-
H. Chen, Q.-J. Hong, S. Ushakov, A. Navrotzky, and A. van de Walle.
A simple method for computing the formation energies of metal oxides.
Comput. Mat. Sci., 198:110692, 2021.
Download: [DOI]
[Cite]
.
- 17
-
S. Zhu and A. van de Walle.
Computational assessment of novel predicted compounds in the Ni-Re
alloy system.
J. of Phase Equilibria, 42:315, 2021.
Download: [DOI]
[Cite]
[NSF
PAR].
- 18
-
S. Samanta and A. van de Walle.
Rapid screening of high-throughput ground state predictions.
Calphad, 74:102306, 2021.
Download: [DOI]
[Cite]
.
- 19
-
C. Nataraj, A. van de Walle, and A. Samanta.
Temperature-dependent configurational entropy calculations for
refractory high-entropy alloys.
Journal of Phase Equilibria and Diffusion, 2021.
Download: [DOI]
[Cite]
[NSF
PAR].
- 20
-
Q. Hong, J. Schroer, D. Hofmann, S. Curtarolo, M. Asta, and A. van de Walle.
Theoretical prediction of melting temperature for a Mo-Ru-Ta-W
HCP multi-principal element alloy.
NPJ Computational Materials, 7:1, 2021.
Download: [DOI]
[Cite]
[NSF
PAR].
- 21
-
H. Liu, M. Asta, and A. van de Walle.
Computational assessment of the efficacy of oxidation-resistant
iridium coatings for multiple principal component rhenium substitutes.
Scripta Mater., 189:16, 2020.
Download: [DOI]
[Cite]
.
- 22
-
Q.-J. Hong and A. van de Walle.
Re-entrant melting of sodium, magnesium and aluminum: General
trend.
Phys. Rev. B Rapid Communications, 100:140102(R), 2019.
Download: [DOI]
[Cite]
[arXiv]
[NSF
PAR].
- 23
-
S. V. Ushakov, A. Navrotsky, Q.-J. Hong, and A. van de Walle.
Carbides and nitrides of zirconium and hafnium.
Materials, 12:2728, 2019.
Download: [DOI]
[Cite]
[NSF
PAR].
- 24
-
R. Sun, M. Asta, and A. van de Walle.
First-principles thermal compatibility between Ru-based
Re-substitute alloys and Ir coatings.
Comp. Mater. Sci., 170:109199, 2019.
Download: [DOI]
[Cite]
.
- 25
-
A. van de Walle, J. Sabisch, A. M. Minor, and M. D. Asta.
Identifying rhenium substitute candidate
multi-principal-element alloys from electronic structure and thermodynamic
criteria.
Journal of Materials Research, 34:3296, 2019.
Download: [DOI]
[Cite]
[Full
text].
- 26
-
A. van de Walle and M. Asta.
High-throughput calculations in the context of alloy design.
MRS Bull., 44:252, 2019.
Download: [DOI]
[Cite]
.
- 27
-
A. van de Walle and Q.-J. Hong.
Assessing phase diagram accuracy.
J. Phase Equilib. Diff., 40:170, 2019.
Download: [DOI]
[Cite]
.
- 28
-
S. Kadkhodaei and A. van de Walle.
A simple local expression for the prefactor in transition state
theory.
J. Chem. Phys., 150:144105, 2019.
Download:
[arXiv]
[DOI]
[Cite]
.
- 29
-
S. Kadkhodaei and A. van de Walle.
Software tools for thermodynamic calculation of mechanically unstable
phases from first-principles data.
Comput. Phys. Commun., 246:106712, 2019.
Download: [DOI]
[Cite]
[NSF
PAR].
- 30
-
M. Fyhrie, Q. Hong, D. Kapush, S.V. Ushakov, H. Liu, A. van de Walle, and
A. Navrotsky.
Energetics of melting of Yb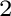 O and LuO from drop
and catch calorimetry and first principles computations.
O and LuO from drop
and catch calorimetry and first principles computations.
The Journal of Chemical Thermodynamics, 132:405, 2019.
Download: [DOI]
[Cite]
[NSF
PAR].
- 31
-
Q.-J. Hong, S. V. Ushakov, D. Kapush, C. J. Benmore, R. J. K. Weber, A. van de
Walle, and A. Navrotsky.
Combined computational and experimental investigation of high
temperature thermodynamics and structure of cubic ZrO and HfO.
Sci. Rep., 8:14962, 2018.
Download: [DOI]
[Cite]
.
- 32
-
A. van de Walle, C. Nataraj, and Z.-K Liu.
The thermodynamic database database.
Calphad, 61:173, 2018.
Download: [DOI]
[Cite]
.
- 33
-
A. van de Walle.
Reconciling SGTE and ab initio enthalpies of the elements.
Calphad, 60:1, 2018.
Download: [DOI]
[Cite]
.
- 34
-
S. Kadkhodaei and A. van de Walle.
Free energy calculations of the mechanically unstable phases of
PtTi and NiTi.
Acta Mater., 147:296, 2018.
Download: [DOI]
[Cite]
.
- 35
-
A. van de Walle, R. Sun, Q.-J. Hong, and S. Kadkhodaei.
Software tools for high-throughput calphad from first-principles
data.
Calphad, 58:70, 2017.
Download: [DOI]
[Cite]
.
- 36
-
Q.-J. Hong, J. Yasi, and A. van de Walle.
A tetrahedron-tiling method for crystal structure prediction.
Phys. Rev. Materials Rapid Communications, 1:020801(R), 2017.
Download: [DOI]
[Cite]
[arXiv].
- 37
-
A. van de Walle, S. Kadkhodaei, R. Sun, and Q.-J. Hong.
Epicycle method for elasticity limit calculations.
Phys. Rev. B, 95:144113, 2017.
Download: [DOI]
[Cite]
[arXiv].
- 38
-
R. Sun, C. Woodward, and A. van de Walle.
First-principles study on NiAl 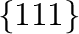 antiphase boundaries
with Ti and Hf impurities.
antiphase boundaries
with Ti and Hf impurities.
Phys. Rev. B, 95:214121, 2017.
Download: [DOI]
[Cite]
.
- 39
-
D. Kapush, S. V. Ushakov, A. Navrotsky, Q.-J. Hong, H. Liu, and A. van de
Walle.
A combined experimental and theoretical study of enthalpy of phase
transition and fusion of yttria above 2000 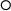 C using 'drop-n-catch'
calorimetry and first-principles calculations.
C using 'drop-n-catch'
calorimetry and first-principles calculations.
Acta Mater., 124:204, 2017.
Download: [DOI]
[Cite]
.
- 40
-
S. Kadkhodaei, Q.-J. Hong, and A. van de Walle.
Free energy calculation of mechanically unstable but dynamically
stabilized bcc titanium.
Phys. Rev. B, 95:064101, 2017.
Download: [DOI]
[Cite]
[arXiv]
[Featured in
Kaleidoscope].
- 41
-
R. Chinnappan, B. K. Panigrahi, and A. van de Walle.
First-principles study of phase equilibrium in Ti-V, Ti-Nb, and
Ti-Ta alloys.
Calphad, 54:125, 2016.
Download: [DOI]
[Cite]
.
- 42
-
M. de Jong, L. Qi, D. L. Olmsted, A. van de Walle, and M. Asta.
Calculations of planar defect energies in substitutional alloys using
the special-quasirandom-structure approach.
Phys. Rev. B, 93:094101, 2016.
Download: [DOI]
[Cite]
.
- 43
-
R. Sun and A. van de Walle.
Automating impurity-enhanced antiphase boundary energy calculations
from ab initio monte carlo.
Calphad, 53:20, 2016.
Download: [DOI]
[Cite]
.
- 44
-
Q.-J. Hong and A. van de Walle.
A user guide for SLUSCHI (solid and liquid in ultra small
coexistence with hovering interfaces).
Calphad, 52:88, 2016.
Download: [DOI]
[Cite]
.
- 45
-
P. Tiwary and A. van de Walle.
A review of enhanced sampling approaches for accelerated molecular
dynamics.
In G. Tucker and C. Weinberger, editors, Multiscale materials
modeling for nanomechanics. Springer, 2016.
Download: [DOI]
[Cite]
.
- 46
-
A. van de Walle, Q.-J. Hong, S. Kadkhodaei, and R. Sun.
The free energy of mechanically unstable phases.
Nature Commun., 6:7559, 2015.
Download: [DOI]
[Cite]
.
- 47
-
Q.-J. Hong and A. van de Walle.
Prediction of the material with highest known melting point from ab
initio molecular dynamics calculations.
Phys. Rev. B Rapid Communications, 92:020104(R), 2015.
Download: [DOI]
[Cite]
(Editor's Suggestion).
Featured in the Washington
Post
and on the Brown news
site.
- 48
-
M. M. de Jong, J. Kacher, M.H.F. Sluiter, L. Qi, D.L. Olmsted, A. van de Walle,
J. W. Morris, A.M. Minor, and M. D. Asta.
Electronic origins of anomalous twinning in hexagonal close packed
transition metals.
Phys. Rev. Lett., 115:065501, 2015.
Download: [DOI]
[Cite]
Featured on the
cover.
- 49
-
L. Miljacic, S. Demers, Q.-J. Hong, and A. van de Walle.
Equation of state of solid, liquid and gaseous tantalum from first
principles.
Calphad, 51:133, 2015.
Download: [DOI]
[Cite]
.
- 50
-
Q.-J. Hong, S. V. Ushakov, A. Navrotsky, and A. van de Walle.
Combined computational and experimental investigation of the
refractory properties of LaZrO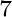 .
.
Acta Mater., 84:275-282, 2015.
Download: [DOI]
[Cite]
.
- 51
-
A. van de Walle.
Simulations provide a rare look at real melting (in
“perspectives”).
Science, 346:704, 2014.
Download: [DOI]
[Cite]
[Full
text].
- 52
-
C. Woodward, A. van de Walle, M. D. Asta, and D. Trinkle.
First-principles study of interfacial boundaries in Ni-NiAl.
Acta Mater., 75:60-70, 2014.
Download: [DOI]
[Cite]
.
- 53
-
A. van de Walle, B. G. Chirranjeevi, S. Demers, Q.-J. Hong, A. Kowalski,
L. Miljacic, G. S. Pomrehn, and P. Tiwary.
Ab initio calculation of anisotropic interfacial excess free
energies.
Phys. Rev. B, 89:184101, 2014.
Download: [DOI]
[Cite]
[arXiv]
[Featured in
Kaleidoscope].
- 54
-
P. Dalach, D. E. Ellis, and A. van de Walle.
Adaptive cluster expansions and redox-dependent atomic ordering.
Comp. Mater. Sci., 83:207, 2014.
Download: [DOI]
[Cite]
.
- 55
-
G. S. Pomrehn, A. Zevalkink, A. van de Walle, and G. J. Snyder.
Defect-driven properties in AZnSb Zintl phases (A=Ca,
Sr, Eu, Yb).
Angew. Chem. Int. Ed., 53, 2014.
Download: [DOI]
[Cite]
.
- 56
-
A. van de Walle.
Methods for first-principles alloy thermodynamics.
JOM - J. Min. Met. Mat. S., 65:1523-1532, 2013.
Download: [DOI]
[Cite]
.
- 57
-
A. van de Walle, P. Tiwary, M. M. de Jong, D. L. Olmsted, M. D. Asta, A. Dick,
D. Shin, Y. Wang, L.-Q. Chen, and Z.-K. Liu.
Efficient stochastic generation of special quasirandom structures.
Calphad, 42:13-18, 2013.
Download: [DOI]
[Cite]
.
- 58
-
Q.-J. Hong and A. van de Walle.
Solid-liquid coexistence in small systems: A statistical method to
calculate melting temperatures.
J. Chem. Phys., 139:094114, 2013.
Download: [DOI]
[Cite]
.
- 59
-
P. Tiwary and A. van de Walle.
Accelerated molecular dynamics simulations through stochastic
iterations and collective variable based basin identification.
Phys. Rev. B, 87:094304, 2013.
Download: [DOI]
[Cite]
[arXiv].
- 60
-
M. M. de Jong, D. L. Olmsted, A. van de Walle, and M. D. Asta.
First-principles study of the structural and elastic properties of
rhenium-based transition-metal alloys.
Phys. Rev. B, 86:224101, 2012.
Download: [DOI]
[Cite]
.
- 61
-
S. Demers and A. van de Walle.
Intrinsic defects and dopability of zinc phosphide.
Phys. Rev. B, 85:195208, 2012.
Download: [DOI]
[Cite]
.
- 62
-
B. G. Chirranjeevi and A. van de Walle.
Ab initio thermodynamics of intrinsic oxygen vacancies in ceria.
Phys. Rev. B, 86:134117, 2012.
Download: [DOI]
[Cite]
[arXiv].
- 63
-
Q.-J. Hong and A. van de Walle.
Direct first-principles chemical potential calculations of liquids.
J. Chem. Phys., 137:094114, 2012.
Download: [DOI]
[Cite]
.
- 64
-
P. Dalach, D. E. Ellis, and A. van de Walle.
First-principles thermodynamic modeling of lanthanum chromate
perovskites.
Phys. Rev. B, 85:014108, 2012.
Download: [DOI]
[Cite]
.
- 65
-
C. Ravi, B. K. Panigrahi, M. C. Valsakumar, and A. van de Walle.
First-principles calculation of phase equilibrium of V-Nb, V-Ta,
and Nb-Ta alloys.
Phys. Rev. B, 85:054202, 2012.
Download: [DOI]
[Cite]
.
- 66
-
B. P. Burton and A. van de Walle.
First principles phase diagram calculations for the
octahedral-interstitial system HfO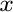 ,
,
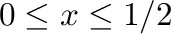 .
.
Calphad, 37:151-157, 2012.
Download: [DOI]
[Cite]
[arXiv].
- 67
-
L. G. Wang and A. van de Walle.
Ab initio calculations of the melting temperatures of refractory bcc
metals.
Phys. Chem. Chem. Phys., 14:1529, 2012.
Download: [DOI]
[Cite]
.
- 68
-
B. P. Burton, A. van de Walle, and H. T. Stokes.
First principles phase diagram calculations for the
octahedral-interstitial system ZrO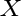 ,
,
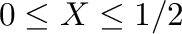 .
.
J. Phys. Soc. Jpn., 81:014004, 2012.
Download: [DOI]
[Cite]
[arXiv].
- 69
-
P. Tiwary and A. van de Walle.
Hybrid deterministic and stochastic approach for efficient long time
scale atomistic simulations.
Phys. Rev. B, 84:100301(R), 2011.
Download: [DOI]
[Cite]
[arXiv].
- 70
-
L. G. Wang, A. van de Walle, and D. Alfè.
Melting temperature of tungsten from two ab initio approaches.
Phys. Rev. B, 84:092102, 2011.
Download: [DOI]
[Cite]
.
- 71
-
G. S. Pomrehn, E. S. Toberer, G. J. Snyder, and A. van de Walle.
Predicted electronic and thermodynamic properties of a newly
discovered Zn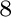 Sb phase.
Sb phase.
J. Am. Chem. Soc., 133:11255, 2011.
Download: [DOI]
[Cite]
.
- 72
-
B. P. Burton, S. Demers, and A. van de Walle.
First principles phase diagram calculations for the
wurtzite-structure quasibinary systems SiC-AlN, SiC-GaN and SiC-InN.
J. Appl. Phys., 110:023507, 2011.
Download: [DOI]
[Cite]
.
- 73
-
G. S. Pomrehn, E. S. Toberer, G. J. Snyder, and A. van de Walle.
Entropic stabilization and retrograde solubility in Zn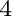 Sb.
Sb.
Phys. Rev. B, 83:094106, 2011.
Download: [DOI]
[Cite]
.
- 74
-
P. Tiwary, A. van de Walle, B. Jeon, and N. Gronbech-Jensen.
Interatomic potentials for mixed oxide and advanced nuclear fuels.
Phys. Rev. B, 83:094104, 2011.
Download: [DOI]
[Cite]
[arXiv].
- 75
-
B. Meredig, A. Thompson, H.A. Hansen, C. Wolverton, and A. van de Walle.
A method for locating low-energy solutions within DFT+U.
Phys. Rev. B, 82:195128, 2010.
Download: [DOI]
[Cite]
.
- 76
-
P. Dalach, D. E. Ellis, and A. van de Walle.
First principles thermodynamic modeling of atomic ordering in
yttria-stabilized zirconia.
Phys. Rev. B, 82:144117, 2010.
Download: [DOI]
[Cite]
.
- 77
-
V.L. Vinograd, N. Paulsen, B. Winkler, and A. van de Walle.
Thermodynamics of mixing in the ternary rhombohedral carbonate solid
solution, (Ca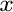 Mg
Mg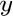 Mn
Mn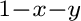 )CO, from atomistic simulations.
)CO, from atomistic simulations.
Calphad, 34:113, 2010.
Download: [DOI]
[Cite]
.
- 78
-
E. Cockayne and A. van de Walle.
Building effective models from scarce but accurate data: Application
to an alloy cluster expansion model.
Phys. Rev. B, 81:012104, 2010.
Download: [DOI]
[Cite]
[arXiv].
- 79
-
C. Ravi, A. van de Walle, B. K. Panigrahi, H. K. Sahu, and M. C. Valsakumar.
Cluster expansion-monte carlo study of phase stability of vanadium
nitrides.
Phys. Rev. B, 81:104111, 2010.
Download: [DOI]
[Cite]
.
- 80
-
A. van de Walle.
First-principles alloy thermodynamics.
In P. Derosa and T. Cagin, editors, Multiscale Modeling: From
Atoms to Devices. CRC press, 2010.
- 81
-
R. V. Chepulskii, W. H. Butler, A. van de Walle, and S. Curtarolo.
Surface segregation in nanoparticles from first principles: The case
of FePt.
Scripta Materialia, 62:179, 2010.
Download: [DOI]
[Cite]
[arXiv].
- 82
-
P. Tiwary, A. van de Walle, and N. Gronbech Jensen.
Ab initio construction of interatomic potentials for uranium dioxide
across all interatomic distances.
Phys. Rev. B, 80:174302, 2009.
Download: [DOI]
[Cite]
[arXiv].
- 83
-
A. van de Walle.
Multicomponent multisublattice alloys, nonconfigurational
entropy and other additions to the Alloy Theoretic Automated Toolkit.
Calphad, 33:266-278, 2009.
Download: [DOI]
[Cite]
[arXiv].
- 84
-
R. Benedek, M. M. Thackeray, and A. van de Walle.
Pourbaix-like phase diagram for lithium manganese spinels in acid.
J. Mat. Chem., 20:369, 2009.
Download: [DOI]
[Cite]
.
- 85
-
O. Adjaoud, G. Steinle-Neumann, B.P. Burton, and A. van de Walle.
First-principles phase diagram calculations for the HfC-TiC,
ZrC-TiC, and HfC-ZrC solid solutions.
Phys. Rev. B, 80:134112, 2009.
Download: [DOI]
[Cite]
.
- 86
-
A. van de Walle.
A complete representation of structure-property relationships in
crystals.
Nat. Mater., 7:455-458, 2008.
Download: [DOI]
[Cite]
[Featured on
cover]
[See associated “News and Views” piece by Gus
Hart].
- 87
-
R. Benedek and A. van de Walle.
Reaction free energies of acid attack of lithium cobaltate.
Journal of the Electrochemical Society, 155:A711, 2008.
Download: [DOI]
[Cite]
.
- 88
-
R. Benedek, M. M. Thackeray, and A. van de Walle.
Free energy for protonation reaction in lithium-ion battery cathode
materials.
Chem. Mater., 20:5485, 2008.
Download: [DOI]
[Cite]
.
- 89
-
G. Ghosh, A. van de Walle, and M. D. Asta.
First-principles calculations of properties of bcc, fcc and hcp solid
solutions in Al-TM (TM = Ti, Zr, Hf) systems: A comparison between
cluster expansion and supercell methods.
Acta Mater., 56:3202, 2008.
Download: [DOI]
[Cite]
.
- 90
-
A. van de Walle and D. Ellis.
First-principles thermodynamics of coherent interfaces in
samarium-doped ceria nanoscale superlattices.
Phys. Rev. Lett., 98:266101, 2007.
Download: [DOI]
[Cite]
.
- 91
-
D. Shin, A. van de Walle, Y. Wang, and Z.-K. Liu.
First-principles study of ternary fcc solution phases from special
quasirandom structures.
Phys. Rev. B, 76:144204, 2007.
Download: [DOI]
[Cite]
[arXiv]
[from PRB
online].
- 92
-
C. H. Lanier, A. van de Walle, N. Erdman, E. Landree, O. Warschkow,
A. Kazimirov, K. R. Poeppelmeier, J. Zegenhagen, M. D. Asta, and L. D. Marks.
The c(6x2) reconstruction on the SrTiO (001) surface.
Phys. Rev. B, 76:045421, 2007.
Download: [DOI]
[Cite]
[arXiv].
- 93
-
J. Z. Liu, G. Ghosh, A. van de Walle, and M. D. Asta.
Transferable force-constant modeling of vibrational thermodynamic
properties in fcc-based Al-TM (TM = Ti, Zr, Hf) alloys.
Phys. Rev. B, 75:104117, 2007.
Download: [DOI]
[Cite]
.
- 94
-
G. Ghosh, A. van de Walle, and M. D. Asta.
First-principles phase stability calculations of pseudobinary alloys
of (Al,Zn)Ti with L1, DO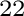 and DO
and DO structures.
structures.
J. Phase Equilib. Diff., 28:9, 2007.
Download: [DOI]
[Cite]
[PDF].
- 95
-
B. Burton, A. van de Walle, and U. Kattner.
First principles phase diagram calculations for the
wurtzite-structure systems AlN-GaN, AlN-InN and GaN-InN.
J. Appl. Phys., 100:113528, 2006.
Download: [DOI]
[Cite]
.
- 96
-
D. Shin, R. Arroyave, Z.-K. Liu, and A. van de Walle.
Thermodynamic properties of binary hcp solution phases from special
quasirandom structures.
Phys. Rev. B, 74:024204, 2006.
Download: [DOI]
[Cite]
.
- 97
-
R. Arroyave, A. van de Walle, and Z.-K. Liu.
First-principles calculations of the Zn-Zr system.
Acta Mater., 54:473, 2006.
Download: [DOI]
[Cite]
.
- 98
-
B.P. Burton and A. van de Walle.
First principles phase diagram calculations for the system
NaCl-KCl: the role of excess vibrational entropy.
Chem. Geol., 225:222, 2006.
Download: [DOI]
[Cite]
.
- 99
-
A. van de Walle.
Genesis of crystal structures (in “news and views”).
Nat. Mater., 4:362, 2005.
Download: [DOI]
[Cite]
.
- 100
-
A. van de Walle, G. Ghosh, and M. D. Asta.
Ab initio modeling of alloy phase equilibria.
In G. Bozzolo, R.D. Noebe, and P. Abel, editors, Applied
Computational Materials Modeling: Theory, Simulation and Experiment. Kluwer
Academic Publishers, 2005.
Download: [DOI]
[Cite]
.
- 101
-
A. van de Walle and M. D. Asta.
First-principles modeling of phase equilibria.
In S. Yip, editor, Handbook of Materials Modeling, volume Part
A. Springer, Dordrecht, the Netherlands, 2005.
Download: [DOI]
[Cite]
[From
Springer].
- 102
-
J. Z. Liu, A. van de Walle, G. Ghosh, and M. D. Asta.
Structure, energetics, and mechanical stability of Fe-Cu bcc alloys
from first-principles calculations.
Phys. Rev. B, 72:144109, 2005.
Download: [DOI]
[Cite]
.
- 103
-
R. Benedek, A. van de Walle, S. Gerstl, M. D. Asta, D. N. Seidman, and
C. Woodward.
Partitioning of solutes in multiphase TiAl alloys.
Phys. Rev. B, 71:094201, 2005.
Download: [DOI]
[Cite]
[from PRB
online].
- 104
-
A. van de Walle, Z. Moser, and W. Gasior.
First-principles calculation of the Cu-Li phase diagram.
Arch. Metall. Mater., 49:535, 2004.
Download: [PDF]
[Caltech].
- 105
-
M. J. Beck, A. van de Walle, and M. D. Asta.
Surface energetics and structure of the Ge wetting layer on Si
(100).
Phys. Rev. B, 70:205337, 2004.
Download: [DOI]
[Cite]
[from PRB
online].
- 106
-
A. van de Walle, M. D. Asta, and P. W. Voorhees.
First-principles calculation of the effect of strain on the diffusion
of Ge adatoms on Si and Ge (001) surfaces.
Phys. Rev. B, 67:041308(R), 2003.
Download: [DOI]
[Cite]
[arXiv]
[from PRB
online].
- 107
-
E. Wu, G. Ceder, and A. van de Walle.
Using bond-length-dependent transferable force constants to predict
vibrational entropies in Au-Cu, Au-Pd, and Cu-Pd alloys.
Phys. Rev. B, 67:134103, 2003.
Download: [DOI]
[Cite]
[arXiv].
- 108
-
B. Burton and A. van de Walle.
First-principles-based calculations of the CaCO-MgCO and
CdCO-MgCO subsolidus phase diagrams.
Phys. Chem. Miner., 30:88, 2003.
Download: [DOI]
[Cite]
.
- 109
-
D. Morgan, B. Wang, G. Ceder, and A. van de Walle.
First-principles study of magnetism in spinel MnO.
Phys. Rev. B, 67:134404, 2003.
Download: [DOI]
[Cite]
[From PRB
online].
- 110
-
D. Balachandran, D. Morgan, G. Ceder, and A. van de Walle.
First-principles study of the structure of stoichiometric and
Mn-deficient MnO.
J. of Solid State Chem., 173:462, 2003.
Download: [DOI]
[Cite]
.
- 111
-
A. van de Walle and G. Ceder.
The effect of lattice vibrations on substitutional alloy
thermodynamics.
Rev. Mod. Phys., 74:11-45, 2002.
Download: [DOI]
[Cite]
[arXiv]
[errata].
- 112
-
A. van de Walle, M. D. Asta, and G. Ceder.
The Alloy Theoretic Automated Toolkit: A user guide.
Calphad, 26:539-553, 2002.
Download: [DOI]
[Cite]
[arXiv].
- 113
-
A. van de Walle and G. Ceder.
Automating first-principles phase diagram calculations.
J. Phase Equilib., 23:348-359, 2002.
Download: [DOI]
[Cite]
[arXiv]
[PDF 1173 KB]
[PDF 2701 KB]
[paper errata (preprint ok)].
- 114
-
A. van de Walle and M. D. Asta.
Self-driven lattice-model monte carlo simulations of alloy
thermodynamic properties and phase diagrams.
Model. Simul. Mater. Sc., 10:521, 2002.
Download: [DOI]
[Cite]
[arXiv].
- 115
-
A. van de Walle and M. D. Asta.
First-principle investigation of perfect and diffuse anti-phase
boundaries in hcp-based Ti-Al alloys.
Metallurgical and Materials Transactions A, 33A:735, 2002.
Download: [DOI]
[Cite]
[arXiv]
[from Science
Direct].
- 116
-
G. Ghosh, A. van de Walle, M. D. Asta, and G.B. Olson.
Phase stability of the Hf-Nb system: From first-principles to
CALPHAD.
Calphad, 26:491, 2002.
Download: [DOI]
[Cite]
.
- 117
-
D. Morgan, D. Balachandran, G. Ceder, and A. van de Walle.
A drastic influence of point defects on phase stability in MnO.
In M. Greenblatt, M.A. Alario-Franco, M.S. Whittingham, and
G. Rohrer, editors, MRS Proceedings, volume 755, pages DD2.8-1, 2002.
Download: [DOI]
[Cite]
[From
MRS site].
- 118
-
H. Ramalingam, M. D. Asta, A. van de Walle, and J. J. Hoyt.
Atomic-scale simulation study of equilibrium solute adsorption at
alloy solid-liquid interfaces.
Interface Science, 10:149, 2002.
Download: [DOI]
[Cite]
.
- 119
-
A. van de Walle and G. Ceder.
First-principles computation of the vibrational entropy of ordered
and disordered PdV.
Phys. Rev. B, 61:5972, 2000.
Download: [DOI]
[Cite]
[arXiv].
- 120
-
D. Morgan, A. van de Walle, G. Ceder, J. D. Althoff, and D. de Fontaine.
Vibrational thermodynamics: coupling of chemical order and size
effects.
Modelling Simul. Mater Sci Eng., 8:295, 2000.
Download: [DOI]
[Cite]
[PDF].
- 121
-
A. van de Walle and G. Ceder.
Correcting overbinding in LDA calculations.
Phys. Rev. B, 59:14992, 1999.
Download: [DOI]
[Cite]
.
- 122
-
A. van de Walle, G. Ceder, and U. V. Waghmare.
First-principles computation of the vibrational entropy of ordered
and disordered NiAl.
Phys. Rev. Lett., 80:4911, 1998.
Download: [DOI]
[Cite]
[arXiv].
- 123
-
A. van de Walle, C. Tricot, and M. Gerspacher.
Modeling carbon black reinforcement in rubber compound.
Kautschuk Gummi Kunststoffe, 49:172, 1996.
Download: [gzipped PS]
[PDF].
BiBTeX file of all publications
[email protected] Sun, Oct 29, 2023 3:23:48 PM