Finding high-coverage configurations
(or: Constrained global optimization)
by Andrew Peterson
Often in catalysis, we can expect coverages of adsorbates under reactive conditions to be quite high. In practice, finding the stable configurations of coverages on a surface can be a combinatorical nightmare, as many degrees of freedom are present. Take the example of a simple 3x3 atom slab in the fcc (211) configuration. To exhaustively search for the lowest energy configuration of just two point adsorbates, we estimate that you would need to manually set up 230 individual calculations. Add in another adsorbate or a rotational degree of freedom, and it quickly becomes unmanageable. This led us to look for automated search methods. This page describes how we conduct such global-optimization searches. This is described here for high-coverage searches, but in principle can be used to look for low-energy structures in any constrained environment.
We like the minima hopping technique, as proposed by Goedecker; our implementation of this in ASE and the technical details of preserving molecular identity were described by me here. This method is nice because it uses standard atomistic tools --- molecular dynamics and local optimizations --- along with self-adjusting parameters to search for minima. The output of such a search is a list of local minima -- or low-energy atomic configurations.
Here, I describe how we automate this procedure to set up several minima hopping jobs in parallel, and direct the jobs to re-submit themselves until the they are finished. If you just want a quick example of the constrained minima hopping method, see the tutorial that I wrote for the ASE project.
Build the identity-preserved atoms
Minima hopping uses high-temperature molecular dynamics, and this can have the effect of breaking chemical bonds. For example, if you are looking for configurations of CO on a surface, you may find that it dissociates and you are instead getting configurations of C and O on the surface. Interesting, but not what is intended. To prevent this, we developed the Hookean constraint in ASE; this constraint applies a restorative force if any specified atom-atom distance exceeds a threshold value. (Note the way in which it does this is conceptually compatible with molecular dynamics: it does not reduce the number of degrees of freedom or violate the conservation of energy. Nor does it fix any bond lengths. Thus it plays well with the minima hopping technique.)
Here, we will look at an example of finding configurations of two COs on a Cu (110) surface. The code below builds the system as you normally would in ASE, but puts in a couple of constraints to preserve identity. We put this in a function to keep the code neat.
from ase import Atoms, Atom
from ase.lattice.surface import fcc110
from ase.constraints import FixAtoms, Hookean
def make_atoms():
constraints = []
atoms = fcc110('Cu', (3, 3, 3), vacuum=12.)
# Build and add the CO adsorbates.
for site in [22, 26]:
ads = Atoms([Atom('C', (0., 0., 0.)),
Atom('O', (0., 0., 1.))])
ads.translate(atoms[site].position + (0., 0., 1.))
atoms.extend(ads)
constraints.append(Hookean(a1=len(atoms) - 1,
a2=len(atoms) - 2,
k=10., rt=1.58))
constraints.append(Hookean(a1=len(atoms) - 2,
a2=(0., 0., 1.,
-(atoms[site].z + 2.2)),
k=10.))
constraints.append(FixAtoms(indices=[atom.index for atom in atoms
if atom.symbol == 'Cu']))
atoms.set_constraint(constraints)
atoms.set_pbc(True)
return atoms
if __name__ == '__main__':
atoms = make_atoms()
from ase.visualize import view
view(atoms)
Save this as build.py (or copy this code into your next script).
This will produce:
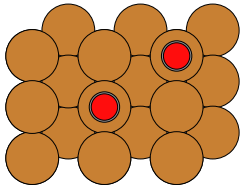
Some things to note:
-
I didn't get concerned about starting with optimized bond lengths in the adsorbate or optimized positioning of it on the slab -- this all will be optimized by the routine.
-
The Hookean constraints were set up only after each adsorbate was added to the atoms object. This is because the indices that are the arguments to this constraint refer to the atoms object.
-
The second Hookean constraint is to prevent volatilization: It keeps the CO from getting too far away from the surface.
Write an auto-restarting script
The minima hopping jobs need to run for a reasonably long time to adjust the parameters and explore the potential energy surface. This will be sped up somewhat by running several jobs in parallel, with all writing to the same minima file.
The below shows an example script.
from ase.optimize.minimahopping import MinimaHopping
from ase.calculators.jacapo import Jacapo
from pgroup.requeue import ReQueue
from build import make_atoms
atoms = make_atoms()
calc = Jacapo(...,
deletenc=True)
atoms.set_calculator(calc)
opt = MinimaHopping(atoms,
minima_traj='../minima.traj',
Ediff0=2.5,
T0=2000.)
requeue = ReQueue(maxtime=24., checktime=0.5)
# Start the compute-intensive part of the job.
status = requeue(opt, maxtemp=4000.)
# See if it finished or if it timed out, requeue if timed out.
if status == 'time_elapsed':
import os
os.system('sbatch --begin=now+2minutes run.py')
Save this as run.py. Some notes:
-
The
ReQueuemodule is a code we wrote to automatically facilitate re-submitting jobs, available here. The way it works in the script above is that every half hour (checktime) it checks to see if the job is still running or if themaxtimehas elapsed. If either is the case it stops and returnsstatusof eithertime_elapsedorjob_completed, if not it keeps running. -
Set
maxtimeto be just less than the time at which your queuing software will kill the job. In this case, this is for a 25-hour job, we have our script kill itself after about 24 hours. Here we have it set to check our job every 0.5 hours; just be sure that it checks it frequently enough -
We wrap the time-intensive line of the code in the
requeuecommand. This command is fed the callable part of the code along with any arguments. That is, if we were not using requeue we would have writtenopt(maxtemp=4000.); this is what will be called by requeue. -
In this code, we requeue our script if it was killed with the
time_elapsedflag. Here, we show the SLURM command for requeuing. Note that we set the job to not start for a couple of minutes, to make sure the current job can exit in a tidy fashion. You can also put clean-up commands here, for example if you want to delete tmp files created by the calculator. -
We have our minima hopping write to a file in the parent directory with the
minima_trajoption, which will allow us to run may scripts in parallel, all working together and writing to a common output file. -
Use whatever calculator you prefer, but note in the case of Jacapo be sure to turn on the
deletencflag so it does not try to open up an existing nc file.
Submit in parallel
This part is simple -- just make a bunch of subdirectories of your working directory (e.g., called run0, run1, ...), copy run.py and build.py into each directory, and submit each run.py. The system should keep your job running (re-submitting) until it reaches the stopping criteria, which in this case is reaching a search temperature of 4000 K. That's it!
Note the way in which it requeues should be pretty friendly to other users. Since your job re-submits every 24 hours: if there are open computational resources it will use them, but because it will stop every 24 hours it will not block other users for more than a day.
Also, our script analyze_MH_runs can automate copying and submitting jobs, with analyze_MH_runs -c. It submits with the SLURM command sbatch, so if you use a different queuing system you will have to modify that line of the script.
(Optional) Run an analysis script
As long as we are automating our lives, we might as well have an automated analysis script. We have one available called analyze_MH_runs available from our group website. To run it, just use ./analyze_MH_runs. This should give you a text output of the status of each of the sub runs, including how many steps it has taken and its current search temperature, as in
$ ./analyze_MH_runs
Path Steps Accepted minima Search temp (K)
run0 5 4 1652
run1 4 3 1652
run2 4 2 2000
run3 4 1 2420
run4 4 1 2420
run5 3 2 1818
=================================================================
Sum 24 13
This script will also create a PDF which has plots of the progress of each job, and a trajectory that contains all of the found minima sorted from lowest to highest.
If you'd rather build your own script, a couple of tips:
-
You can use the command
MHPlot(fromase.optimize.minimahopping) to create plots of the progress of any MH job, as in:mhplot = MHPlot(rundirectory=path) mhplot.save('%s.pdf' % path) -
We sometimes do the minima hopping search with the slab frozen, then re-relax each of the configurations in the minima script with the top layer relaxed. You can also build this into the analysis script.