PROVIDENCE, R.I. [Brown University] — Every year, fungal infections threaten thousands of patients — from those with depressed immune systems to others who have had surgeries or devices such as catheters implanted. Moreover, some anti-fungal medications are beginning to lose their power.
To help protect patients, a team of scientists at Brown University and Rhode Island Hospital has focused on understanding the body’s natural immune response, revealing key details in a new study that could ultimately help to develop new therapies.
“To combat rising fungal infection rates and growing drug resistance, an increased focus on additional ways to enhance the innate immune and anti-fungal response may offer alternative treatments,” wrote the authors of the study in the Journal of Immunology.
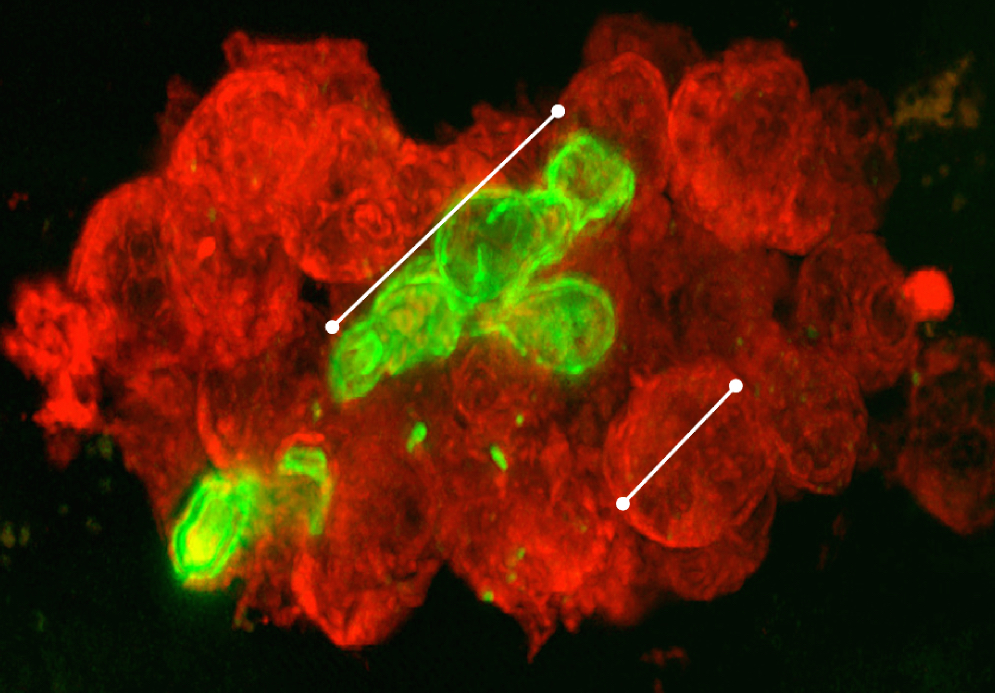
The research reveals how three receptors called integrins on the surface of the white blood cells called neutrophils signal the cells to take action against the fungus as it spreads its tendrilous hyphae into infected tissue.
The research team in the Rhode Island Hospital lab of Jonathan Reichner, a research professor of surgery at Brown’s Alpert Medical School, not only observed these specific mechanisms, but also manipulated the integrins in the lab to change the behavior of the neutrophils. That sets the table for studies in which scientists can gauge the clinical effects of doing so to see if they can improve the body’s defenses.
“We are looking for ways to mobilize and activate our neutrophils to eliminate fungal infections, especially hyphal forms of fungal infections,” said Courtney Johnson, a fourth-year medical student who earned her Ph.D. from Brown who co-led the study with Xian O’Brien. “At the same time, by learning more about how neutrophils cluster and what happens in those aggregations, we can also learn when they may be causing damage to tissue.”
How the battle proceeds
The battle between the immune system and the fungus plays out in the extracellular matrix — the area of tissues that lies between cells. There, patrolling neutrophils from the bloodstream sense the presence of beta-glucan, a part of the fungal cell wall.