PROVIDENCE, R.I. [Brown University] — Researchers from Brown University have developed a new method for sifting through genomic data in search of genetic variants that have helped populations adapt to their environments. The technique, dubbed SWIF(r), could be helpful in piecing together the evolutionary history of people around the world, and in shedding light on the evolutionary roots of certain diseases and medical conditions.
SWIF(r) brings several different statistical tests together into a single machine-learning framework. That framework can then be used to scan genomic data from multiple individuals and compute the probabilities that individual mutations or regions of a genome are adaptive.
“These individual statistical techniques are useful, but none of them is particularly powerful on its own,” said Lauren Alpert Sugden, a postdoctoral researcher at Brown who led the technique’s development. “The method we’ve developed combines those techniques in a way that’s careful and that produces an output that’s easy to interpret.”
Alpert Sugden works in the lab of Sohini Ramachandran, an associate professor and director of Brown’s Center for Computational Molecular Biology. The researchers describe their work in the journal Nature Communications.
Exploring adaptation
The vast majority of mutations that commonly occur in the genomes of humans and other animals are neutral, meaning they neither help nor hurt an individual’s survival. But every once in a while nature hits on a mutation that’s beneficial — one that aids in an organism’s survival or reproductive success. These adaptive mutations can spread quickly (evolutionarily speaking) through a population in subsequent generations, a process known as a selective sweep.
SWIF(r) looks for the statistical signatures of selective sweeps in genomic datasets. It does so using machine learning and a combination of four established statistical tests measuring different signatures of adaptation. One test checks if a particular mutation appears in a population more frequently than it does in other populations. Others measure genetic variation in a region of the genome, with the idea that strong selection would tend to reduce variability.
This isn’t the first technique that brings multiple tests into one composite framework. But part of what’s new about SWIF(r) is that it controls for correlations that arise between those tests, which can throw off the results. The acronym SWIF(r) stands for “SWeep Inference Framework (controlling for correlation),” a lowercase “r” being the mathematical notation for correlation.
SWIF(r) has several advantages over other composite techniques, the researchers say. While most techniques identify only regions of the genome likely to contain adaptive mutations, SWIF(r) can also identify the particular mutations themselves. And while other techniques return results that can be difficult to interpret, SWIF(r) returns a simple probability that an individual mutation or genome region is adaptive.
To show that the technique works, the researchers validated it on a simulated dataset in which known adaptive mutations were included, as well as on canonical adaptive mutations that have been identified in human genomes through multiple molecular experiments. SWIF(r) was shown to outperform both individual statistical techniques and other composite techniques in picking out those adaptive mutations, while producing a lower rate of false positives.
Real-world data
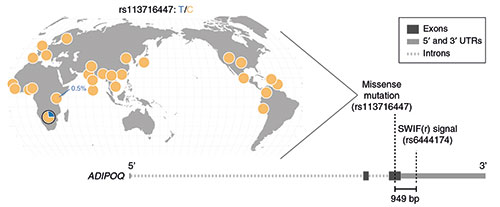
Having demonstrated that SWIF(r) works, the researchers used it on a real genomic data from the ‡Khomani San, a group of hunter-gatherers living in southern Africa.
“The ‡Khomani San have the largest genetic diversity of any living population,” Alpert Sugden said, “which is interesting from our perspective because there’s a lot of opportunity for adaptive mutations to arise.”
Among other findings, SWIF(r) identified several adaptive mutations in a set of genes responsible for energy and fat storage. That’s interesting from the perspective of what’s known as the “thrifty gene” hypothesis, the researchers say.
The hypothesis suggests that because hunter-gatherers often experience an inconsistent food supply, they’re likely to have a genetic predisposition to storing energy in the form of fat. However, those genes could be a liability in agricultural societies where food supply tends to be more consistent, potentially contributing to obesity and complications like type 2 diabetes. A deeper dive into the functions of the adaptive genes identified by SWIF(r) may be helpful in further exploring the thrifty gene idea.
Ramachandran says the way in which they used SWIF(r) on the ‡Khomani San data is instructive for how the technique might be used moving forward. The researchers say they didn’t start with the notion that they’d find adaptations in genes for metabolism, they simply popped out of the data as it was analyzed. That’s a contrast to how such research is currently done, Ramachandran says.
“They way we study genetic adaptation now is we start by looking at a particular trait or phenotype, and then we work backward to identify the associated genes and mutations,” she said. “This new approach uses data-driven machine learning to start in the genome, searching for adaptive signatures that we can then follow up with more study. So we think this is a way of generating new and interesting hypotheses to test.”
The researchers have made the SWIF(r) code open-source, and they hope that other research groups will use it to explore genomic data from populations worldwide.
Additional authors on the paper were Elizabeth G. Atkinson, Annie P. Fischer, Stephen Rong and Brenna M. Henn. The research was supported the National Institutes of Health (R01GM118652, P20GM109035, K12-GM-102778), the National Science Foundation (DBI-1452622, DBI-1452622), the Pew Charitable Trusts and the Alfred P. Sloan Research Foundation.