DNA-grabbing protein flips a calcium channel switch that contributes to chronic pain
Scientists at the Carney Institute for Brain Science have identified one way that a synaptic calcium channel protein in sensory neurons is modulated, providing insight into mechanisms that contribute to chronic pain. The research has the potential to inform new therapeutic targets for abnormal pain conditions.
The two-author study was published in the journal eLife on March 26 and reports work of E. Javier López Soto, postdoctoral research associate at Brown University, and Diane Lipscombe, director of the Carney Institute and a professor of neuroscience at Brown.
Lipscombe has been investigating variations in calcium channel proteins since the 1990s. Calcium ion channels produce electrical signals essential for transmission of information between neurons. Scientists have known that ion channels tend to behave differently depending on the type of cell that houses them.
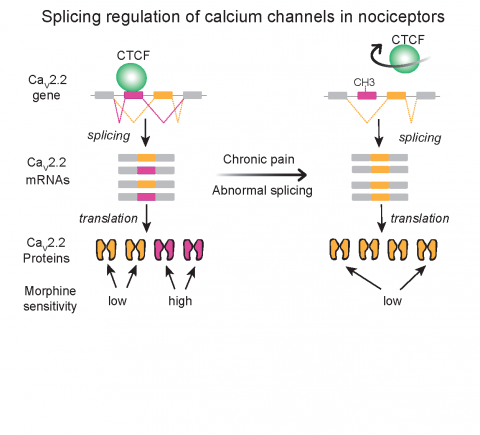
Lipscombe and her colleagues had previously found that a process called RNA splicing controls switching between different types of calcium channels in neurons. This process swaps segments of genetic information, allowing a single gene to produce multiple related but distinct proteins. In particular, they identified splicing events that generate a specific version of a calcium channel called Cav2.2 in neurons that send signals to the brain that cause pain.
Cells containing this version of the Cav2.2 channel are especially sensitive to the pain-killing effects of opioids such as morphine. However, after a nerve injury, cells tend to lose this version of Cav2.2 in favor of one that is less sensitive to morphine. According to López Soto, morphine isn’t an effective painkiller in some chronic pain conditions, and the switch between Cav2.2 versions identified by the researchers could help to explain it.
In the study, López Soto and Lipscombe investigated the molecular mechanism that triggers the switch between the two versions of Cav2.2. By performing cell culture experiments and combing open-source genetic databases, they identified a protein called CTCF that attaches to a crucial segment of the gene encoding Cav2.2.
When bound, this protein encourages production of the morphine-sensitive version of Cav2.2 that functions in healthy pain-signaling neurons. CTCF binding to DNA is, in turn, controlled by patterns of chemical modifications to genomic DNA. The researchers found that nerve injury alters the pattern of chemical modification on DNA, discouraging CTCF from binding and favoring production of the version of Cav2.2 that is more morphine resistant.
“We found the molecular mechanism in cells that is the source of a calcium ion channel with unique properties in pain signaling neurons,” López Soto said.
Now that the researchers have identified the molecular switch between the two versions of the calcium channel, future efforts could include experiments to intervene with the channel’s function, according to Lipscombe. “Could we shift the channel back to its normal state,” she said, “targeting those mechanisms that Javier discovered that are disrupted in chronic pain?”
With support of a 2019 fellowship from the Warren Alpert Foundation, López Soto will be able to pursue new questions prompted by this study. The Lipscombe Lab, he said, is now looking at using next generation sequencing, which allows for high-throughput sequencing of DNA, and other bioinformatics analyses to investigate changes on a genome-wide level.
“My goal now is to better understand the role of these dynamic events, such as DNA binding proteins and methylation, in the onset and maintenance of a neuronal-specific splicing program underlying chronic pain conditions,” López Soto said.



